
Group photo. From left to right. Sol Vendrell, Leticia Lucero, Cristina Fernández, Rafael Giraldo, Silvia Marín
In our research group we address a Synthetic Biology (BS) program whose objective is the development of parts, devices and systems for modeling amyloid proteinopathies in microorganisms (Giraldo, 2010). We thus intend to deconstruct amyloidosis into functional modules whose integration makes their better understanding possible and also, potentially, actions with diagnostic and/or therapeutic purposes. Amyloidoses are degenerative, neuronal or systemic processes of extraordinary complexity from the molecular, cellular and whole organism points of view (Eisenberg and Jucker, 2012). However, in all the cases studied, the causal agent of each type of amyloidosis is relatively simple: a protein, such as Aβ and Tau (in Alzheimer's disease), α-synuclein (in Parkinson's), Htt (in Huntington's) or SOD1 (in ALS), which has at least two conformations; while the native one is soluble, the other alternative aggregates forming fibers with a β-laminar amyloid skeleton. These amyloid aggregates, more specifically their precursor oligomers or those resulting from partial disassembly, are the cytotoxic molecular species. An intriguing property of any amyloid protein is the ability to replicate its aberrant conformation by contact molding it into soluble molecules of that same protein. In reality, the assembled form is composed of a set of metastable conformations whose free energies are very similar, thus being in equilibrium with each other. This is manifested, for a given protein, in the existence of "stocks" or conformational variants and "quasispecies", i.e., families of related conformations subject to selection and replication (Giraldo, 2010; Eisenberg and Jucker, 2012). Amyloid proteins are closely related to prions, as these are a subclass of amyloid but with an additional property that is well known to us as microbiologists: infectivity, that is, their "horizontal" transmissibility, even under experimental conditions, between cells. or entire organisms within a population (Eisenberg and Jucker, 2012). The archetypal prion is PrP, whose various amyloid aggregate conformations are behind the transmissible spongiform encephalopathies in mammals, while the agents of the other amyloidoses are called "prionoids" because they lack infectivity. However, the most recent studies seem to conclude that in all neurodegenerative processes the amyloid conformations follow, in their progressive dispersion through the nervous system, an intercellular propagation of the infective type, which would also allow them to be classified, sensu lato, as prions (Prusiner, 2012).
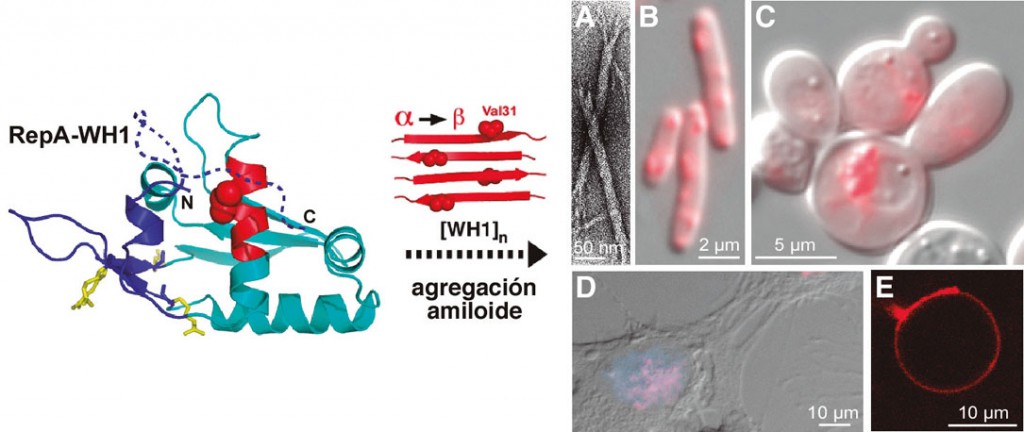
Figure 1. The WH1 domain of the plasmid replication protein RepA (left), upon monomerization, changes part of its a-helical structure into b-strands (in red) and assembles as amyloid fibers (A, electron micrograph). When fused to the fluorescent protein mCherry, RepA-WH1 appears as inclusions in the cytoplasm of E. coli (B) and S. cerevisiae (C), microorganisms in which it acts as a prionoid, as well as when expressing it in the nucleus (blue) of neuroblastoma cells in culture (D). In liposomascytomimetics, RepA-WH1 aggregates in the lipid bilayer (E).
The BS of proteotoxic amyloid modules in microorganisms such as Gram-negative bacteria Escherichia coli or yeast Saccharomyces cerevisiae It is a formidable challenge, since the inherent complexity of protein bioengineering is compounded by the fact that bacteria do not naturally suffer intracellular amyloidosis, while in yeast amyloidosis is not cytotoxic, but a functional resource that confers phenotypic adaptations fast and epigenetically transmissible (White et al., 2012). However, for these same reasons, BS offers us the opportunity to develop microbial models of amyloidosis in which both the etiological agents and their host cells can be conceived. from cognition as orthogonal modules, that is, without prior functional interaction (Giraldo, 2010). When our group was established in 1999, we were interested in studying the structure and mechanism of action of the proteins, called Rep, that initiate DNA replication of bacterial plasmids and, collaterally, the assembly of the ORC replication complex proteins. in S. cerevisiae (Giraldo, 2003). We discovered that the RepA protein encoded by plasmid pPS10 undergoes a conformational change that enables it to go from being a dimer, a transcriptional repressor of the repA gene itself, to being a replication initiator monomer, a process activated by binding to specific DNA sequences that entails a structural remodeling of the N-terminal domain of the protein (WH1) (Giraldo et al, 2003). RepA-WH1 thus increases its b-laminar structure and, finally, the protein adds in a post-application complex that inhibits premature replication restarts (Gasset-Rosa et al, 2008a). In 2007 we determined that RepA-WH1 is assembled in vitro in amyloid fibers in a process promoted by their interaction with oligodeoxyribonucleotides (Giraldo, 2007; Gasset-Rosa et al, 2008b), which led us to explore whether its expression in the bacterium E. coli, uncoupled from plasmid replication, it led to the formation of intracellular amyloid aggregates. Indeed, RepA-WH1, fused to a fluorescent reporter protein, forms cytoplasmic inclusions that are cytotoxic to bacteria and that, once purified, are capable of accelerating amyloid fibrillation in vitro (Fernández-Tresguerres et al., 2010). RepA-WH1, given the absence of homologous proteins in the human proteome in which it could shape its amyloid conformation and its non-infective nature, is classifiable as a biosafety level 1 prionoid. Recently, in collaboration with the group of A. Lindner (INSERM – U. Descartes, Paris) and thanks in part to a FEMS grant, we have studied the “vertical” transmission (from mother cell to daughter cells) of RepAWH1 aggregates using microfluidic techniques. These studies have led us to characterize the existence of two amyloid strains, phenotypically differentiated by their toxicity and the morphology of the aggregates, whose interconversion is dependent on a single cellular factor: DnaK, the chaperone of the Hsp70 family in E. coli (Gasset-Rosa et al., 2014; Molina-Garcia and Giraldo, 2014). Transcriptomic and proteomic approaches still in progress are allowing us to trace, with a clarity that is difficult to achieve in complex cellular systems, the amyloid cytotoxicity pathways. Among the common targets of the proteins involved in human amyloidosis, the plasma membrane stands out, so we are also studying the interaction of RepA-WH1 with liposomes, thus taking the modeling of these processes to the extreme of cytomimetic minimalism. In a diametrically opposite direction, exploring more complex systems, we have been able to establish (Gasset-Rosa and Giraldo, 2014) that the sequence responsible for amyloidosis in RepA-WH1, if arranged in the form of tandem repeats, is capable of substituting in chimeras to those of the Sup35p/[PSI+] yeast prion (Liebman and Chernoff, 2012), thus generating a new functional synthetic prion, which we have named [REP-PSI+]. This manifests itself in the form of various phenotypic variants whose interconversion is regulated, as in its parent RepA-WH1, by a chaperone Hsp70 (Ssa1p) and not by Hsp104p, the chaperone that modulates the propagation of [PSI+] in yeasts ( Liebman and Chernoff, 2012). Finally, we are exploring the expression of RepA-WH1 in cultured human cells and developing systems for screening bacteria for amyloidosis-inhibiting molecules, which will constitute proofs of concept for the potential of synthetic bacterial systems in modeling and pharmacological approach of amyloid proteinopathies with biomedical relevance.
Updated group information can be found at:
Representative bibliography
Blanco LP, Evans ML, Smith DR, Badtke MP y Chapman MR. (2012). Diversity, biogenesis and function of microbial amyloids. Trends Microbiol 20: 66–73.
Eisenberg D y Jucker M. (2012). The amyloid state of proteins in human diseases. Cell 148: 1188–1203.
Fernández-Tresguerres ME, Moreno-Díaz de la Espina S, Gasset-Rosa F and Giraldo R. (2010). A DNA-promoted amyloid proteinopathy in Escherichia coli. Mol Microbiol 77: 1456–1469.
Giraldo R. (2003). Common domains in the initiators of DNA replication in Bacteria, Archaea and Eukarya: Combined structural, functional and phylogenetic perspectives. FEMS Microbiol Rev 26: 533–554.
Giraldo R. (2007). Defined DNA sequences promote the assembly of a bacterial protein into distinct amyloid nanostructures. Proc Natl Acad Sci USA 104: 17388–17393.
Giraldo R. (2010). Amyloid assemblies: Protein legos at a crossroads in bottom-up Synthetic Biology. ChemBioChem 11: 2347–2357.
Giraldo R, Fernández-Tornero C, Evans PR, Díaz-Orejas R and Romero A. (2003). A conformational switch between transcriptional repression and replication initiation in the RepA dimerization domain. Nat Struct Biol 10: 565–571.
Gasset-Rosa F, Díaz-López T, Lurz R, Prieto A, Fernández-Tresguerres ME and Giraldo R. (2008a). Negative regulation of pPS10 plasmid replication: Origin pairing by zipping-up DNA-bound RepA monomers. Mol Microbiol 68: 560–572.
Gasset-Rosa F, Maté MJ, Dávila-Fajardo C, Bravo J, and Giraldo R. (2008b). Binding of sulphonated indigo derivatives to RepA-WH1 inhibits DNA-induced protein amyloidogenesis Nucleic Acids Res 36: 2249–2256.
Gasset-Rosa F, Coquel AS, Moreno-del Álamo M, Chen P, Song X, Serrano AM, Fernández-Tresguerres ME, Moreno-Díaz de la Espina S, Lindner AB, and Giraldo R. (2014). Direct assessment in bacteria of prionoid propagation and phenotype selection by Hsp70 chaperone. Mol Microbiol 91: 1070–1087.
Gasset-Rosa F and Giraldo R. (2014). The Hsp70 chaperone Ssa1p selects for weak variants of [REP-PSI+], a novel bacterial-yeast hybrid prion. En revisión.
Liebman SW y Chernoff YO. (2012). Prions in yeast. Genetics 191:1041–1072.
Molina-Garcia L and Giraldo R. (2014). Aggregation interplay between variants of the RepA-WH1 prionoid in Escherichia coli. J Bacteriol 196: 2536–2542.
Prusiner SB. (2012). A unifying role for prions in neurodegenerative diseases. Science 336